Comprehensive Analysis of DNA Methylation Data with RnBeads
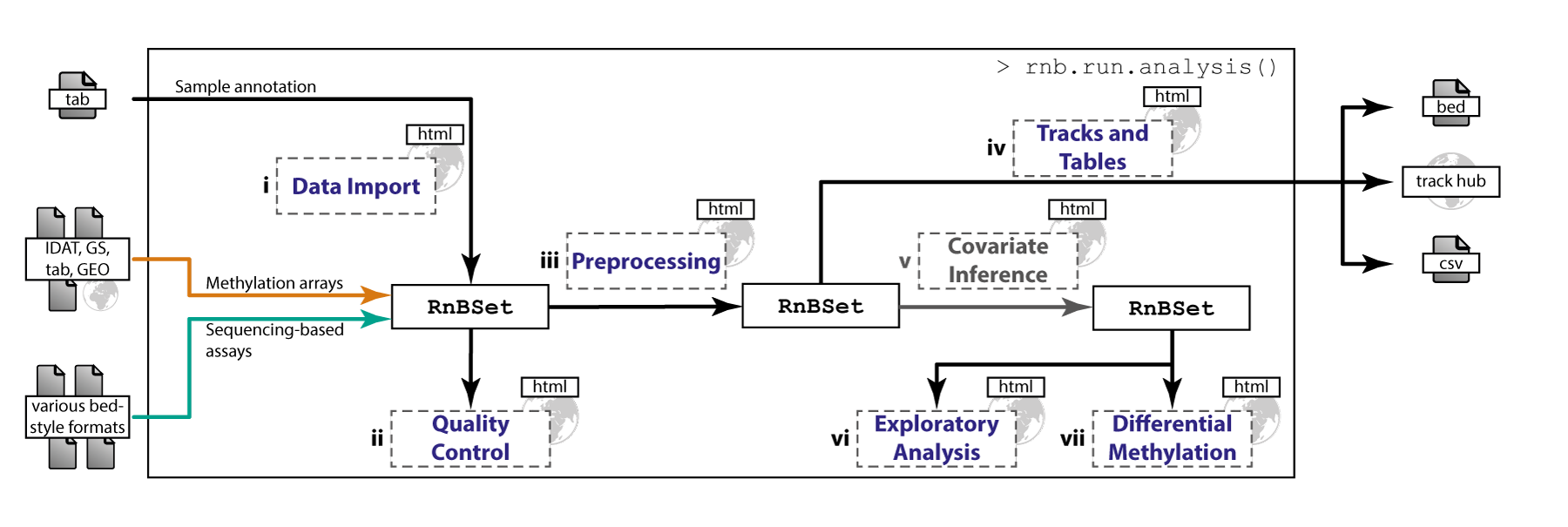
RnBeads is an R package for comprehensive analysis of DNA methylation data obtained with any experimental protocol that provides single-CpG resolution. Supported assays include Infinium and EPIC microarrays and bisulfite sequencing protocols, and also MeDIP-seq and MBD-seq once the data have been preprocessed with DNA methylation level inference software. RnBeads implements an analysis workflow that is significantly more comprehensive than those of existing tools. It documents its results in a highly annotated and readable hypertext report, and it scales to the large sample sizes that are becoming the norm for DNA methylation analysis in human cohorts.
Various DNA methylation assays and input formats are supported. The pipeline implements state-of-the-art normalization techniques. Experimental quality control can be conducted and sample outliers and mix-ups can be identified. The package provides flexible methods for CpG and sample filtering. According to sample annotations, batch effects and phenotype covariates can be identified. DNA methylation distributions are analyzed and intergroup as well as intragroup variability in methylation profiles is quantified. Furthermore, differential methylation between groups of samples can be characterized. The analysis is based on individual CpGs as well as on predefined or custom genomic regions. Finally, methylation data can be exported in various formats including genome browser views. Comprehensive, highly interpretable reports containing method descriptions, publication grade plots and data tables are generated. Their HTML format facilitates easy tracking and comparison of analyses as well as exchanging results with collaboration partners. Due to its modularized concept, both first-time users and experts can conveniently perform analyses according to their individual demands. A single, comprehensive analysis run can be invoked by specifying only few parameters and executing a master command. Alternatively, a user may execute the steps of the pipeline individually.
Update: RnBeads now supports the Illumina mouse array MMBC
Thanks to our collaborators, RnBeads now supports analyzing data generated using the Illumina mouse array MMBC. Check out the preprint describing the first analysis of MMBC data and try out the updated package (development version) on Bioconductor.
Note: RnBeads default options updated
In order to keep up with current practices in DNA methylation analysis, we updated the default options for running the RnBeads pipeline. Of course, all old options are still supported, but not the default anymore. Please refer to the FAQ item for details which options changed.
RnBeads 2.0 published
Happy Pi-day 2019. We are happy to announce that a manuscript describing RnBeads 2.0 has been published in Genome Biology (2019).
New GUI
The new RnBeads version provides a graphical user interface that makes configuring and running RnBeads analysis more accessible to new and experienced users. The GUI vignette and the tutorial page contain detailed descriptions on how to use this powerful tool. In brief, to start the GUI simply load the package and run the command:
library(RnBeads) rnb.run.dj()
Bioconductor
RnBeads is part of Bioconductor. You can install the package and obtain the source code from there.
Publication
A manuscript describing RnBeads 2.0 has been published in Genome Biology (2019). The software was initially published in Nature Methods (2014).
Documentation
The full documentation is available in the package vignette and the reference manual.
Supporting Institutes
RnBeads was developed by the Computational Epigenetics Groups at the Max Planck Institute for Informatics and Saarland University. Its developers now also hold positions at the CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences and the German Cancer Research Center. RnBeads is also supported by the German Network for Bioinformatics Infrastructure (de.NBI).




